Enfermedad de Batten
¿QUÉ ES LA ENFERMEDAD DE BATTEN, O LA CEROIDOLIPOFUSCINOSIS?.
Se trata de un grupo de enfermedades, heredadas con carácter autosómico recesivo, que tienen como dato común el acumulo de un material de depósito en las neuronas, lo que lleva a la degeneración de las mismas y a la subsiguiente sintomatología neurológica, entre la que destaca inicialmente las manifestaciones epilépticas. Se identifican diferentes subgrupos en función de la edad de presentación, poseyendo cada uno de ellos una peculiar característica ultraestructural del lipopigmento neuronal acumulado.La enfermedad de Batten es un desorden hereditario del sistema nervioso que comienza en la niñez. Normalmente, los síntomas iniciales del desorden aparecen entre los 5 y 10 años de edad, cuando los padres o médicos de niños, previamente normales, comienzan a observar que desarrollan problemas de visión y convulsiones. En algunos casos los síntomas iniciales pueden ser repentinos, tomando la forma de cambios en la personalidad y conducta, aprendizaje lento y torpeza, o tropiezos.
¿Cuánta gente padece estos trastornos?
La enfermedad de Batten y otras formas de ceroidolipofuscinosis neuronales son relativamente raras y se dan entre 2 a 4 de cada 100 mil nacimientos en los Estados Unidos. Estos trastornos parecen ser más comunes en Finlandia, Suecia, otras partes de Europa del Norte y en la provincia de Newfoundland en Canadá. Aunque las ceroidolipofuscinosis neuronales se clasifican como enfermedades raras, atacan a menudo a más de una persona en las familias que presentan los genes defectuosos.
Para diagnosticar una ceroidolipofuscinosis neuronal, el neurólogo necesita el historial médico y varias pruebas de laboratorio del paciente. Las pruebas de diagnóstico utilizadas para detectar las ceroidolipofuscinosis neuronal incluyen:
Análisis de sangre o de orina.
Muestras de piel o de tejido.
Electroencefalograma o EEG
Estudios eléctricos de los ojos.
Exploraciones del cerebro.
Medición de la actividad enzimática.
Análisis del ADN.
. 
NO TIENEN TRATAMIENTO !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
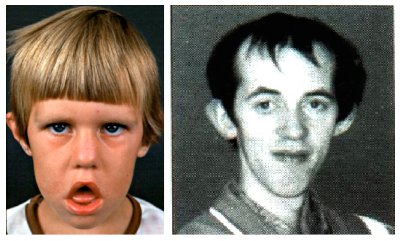
COMO PODEMOS OBSERVAR HAY UNOS SIGNOS CARACTERÍSTICOS Y EVIDENTES EN EL FÍSICO DE LAS PERSONAS QUE SUFREN ESTE TIPO DE ENFERMEDAD.








{kind=link}
{kind=link}